AlphaFold2 has been widely reported as a fantastic leap forward in the prediction of protein structures from sequence, when sequence has enough homologs to build a reasonable multiple sequence alignment. When AlphaFold2 was released (Jumper et al. 2021) there were several independent reports of how it could also be used for the prediction of structures of protein complexes despite the fact that it was not trained to do so (Bryant et al., 2021; Ko and Lee, 2021; Mirdita et al. 2022). Together with the lab of Arne Elofsson, in work led by David Burke in our group and Patrick Bryant in Arne's group, we have shown that it can be applied in reasonably large scale to predict structures of protein complexes for known human interactions (Burke et al. 2021). There is a lot to investigate still but it is clear that this is an extremely exciting direction of research since that lead to a major advances in the structural analysis of cell biology, evolution, biotechnology, etc.
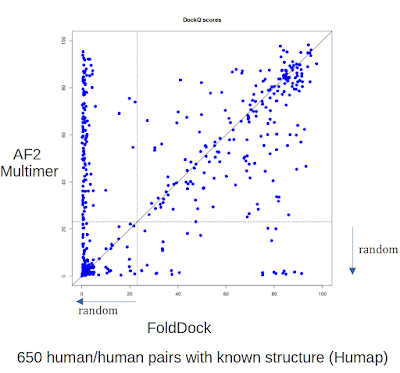
Soon after these first reports, DeepMind released an AlphaFold version that was re-trained specifically for prediction of structures of protein complex - AlphaFold-Multimer (Evans et al. 2021). Given that they reported an even higher success rate with this specifically trained model we were quite excited to give this a try. David Burke selected a set of 650 pairs of human proteins from the Hu.MAP dataset, known to physically interact and for which the experimental structure has been solved. A structure was predicted using AF v2.1.1 (AF-multimer) using default settings and the model_1_multimer parameter set. A second model was predicted using AF using the model1 monomer parameter set and the FoldDock pipeline. For each model, DockQ scores were produced which reflect the similarity of the predicted structure with the experimental structure with a specific focus on the interaction interface residues. A DockQ score value below 0.23 can be considered essentially an incorrect or random model.
Below we show a direct comparison between the two AlphaFold2 models with the AF2 Multimer showing a very significant improvement based on DockQ scores. Of all predictions tested, there were 51% above DockQ>0.23 with AF2 Multimer and 40%>0.23 with "standard" AlphaFold2. This improvement (+11%) is not as large as that reported by the DeepMind team (+25%) on their own test set. There could be several reasons for the difference but more importantly this would be more than enough to justify using Multimer for the prediction of protein complexes.
However, David quickly realised that there were many examples of clashes at the predicted interface with the AF2 Multimer model. In the figure below we show just an example of this which, despite the high DockQ score (0.85) clearly has several overlapping residues. That is, while the interface region is likely to be correct, the model at the interface has serious errors.

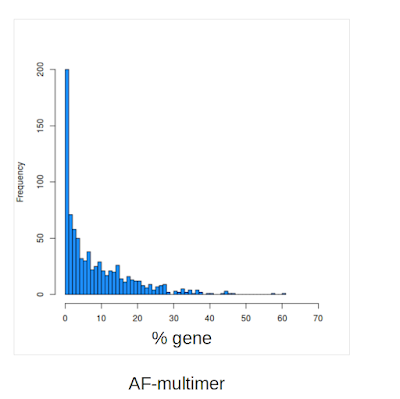
While there may be some cases where the clashes could be minimised, as it stands the models produced by AF-multimer may not be usable for a large fraction of cases. However, these issues are of course easy to spot. DeepMind is in fact aware of this bug since around November and have said they are working on it. From the point of view of predicting the regions of the proteins where the interaction will occur AF-multimer may still be usable as it is and hopefully DeepMind will find a fix for this problem.