As early as in 1975, Mary-Claire King and Allan Wilson observed that there was very little change in many human proteins when compared to their chimp counterparts. From this they postulated the divergence between humans and chimpanzees should be attributed to changes in regulatory regions, that would impact on the expression levels of the proteins.
So, this brings us to one possible consequence of mutations on biological systems. Genomic variation can alter the regulatory networks of the cell, either by mutating transcription factors or by changing the regulatory binding sites where the transcription factors bind to in the genome.
To what extend can the expression change with evolution? One way to gauge for the possible rate of change is to look at recently duplicated proteins pairs within the same species and check how related in expression are they. Several studies (for review see here) have shown that expression divergence occurs quickly after gene duplication. Papp and colleagues provided with a possible mechanism for this change. When they studied the up-stream region of duplicated proteins they saw that the number of shared binding sites decreases with the age of the duplication although the total number of binding sites stays fairly constant.
This provides with a model of change of expression after duplication by quick lost and gain of binding sites by mutations in the regulatory regions.
To determine the impact of divergence on the changes in the regulatory networks one can also look at how conserved are the regulatory regions of the same proteins in different species (orthologs).
Dermitzakis and Clark have calculated that between 32% to 40% of the human transcription factor binding sites are functional in rodents, providing evidence for significant change in regulatory sites.
Gash and colleagues used a different approach to study the cis-regulatory region of S. cerevisiae proteins. They looked at the conservation of binding consensus in several other fungi spanning a very broad evolutionary distance (see figure 1, adapted from the same paper). They have looked at groups of proteins regulated by the same transcription factor and calculated over-represented sequence motifs in their regulatory region. These over-represented motifs likely represent the binding specificity of the transcription factor . They next checked if the same over-represented motif is observed or not in the regulatory regions of orthologous proteins. The result (figure 1) is that the binding consensus is mostly conserved even in species that have diverged from S. cerevisiae a long time ago (~400My). This seems a bit contradictory with the previous results but actually they do not say that the actual biding sites are conserved. My interpretation from their results is that a sufficient number of binding sites are conserved so that we still see an over-representation of the motif. What we can also see is that the binding specificity of the transcription factor is mostly conserved.
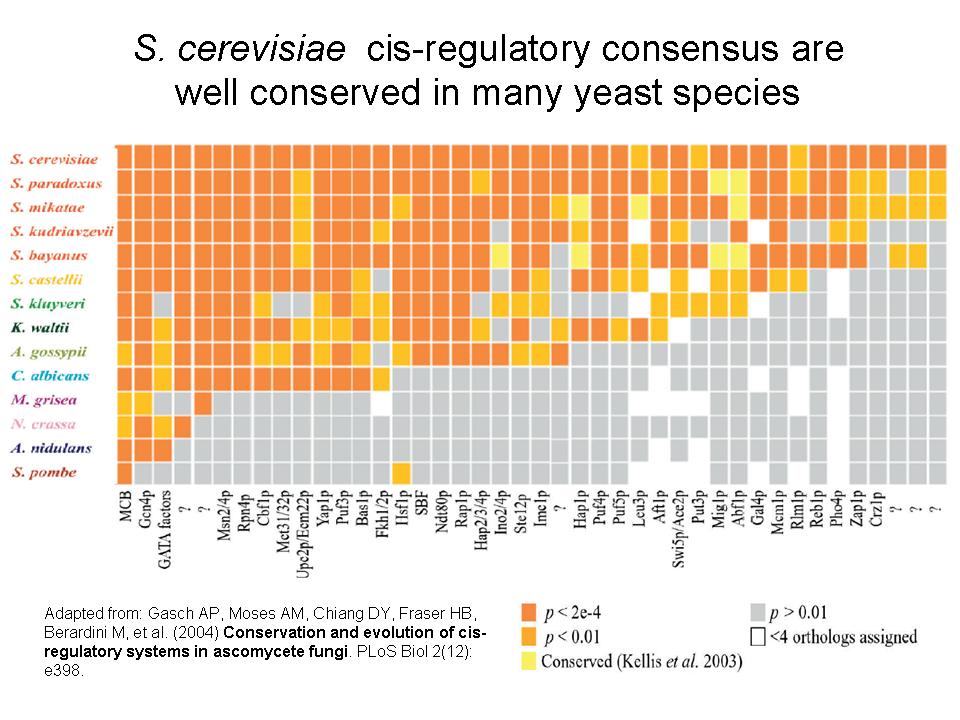
So more generally, what emerges is that expression change occurs mostly through changes in regulatory regions and not so much trough changes in the binding specificity of the transcription factors. To put in in another way, the binding sites are more likely to change then the binding properties of the transcription factors.
These results tell us that genomic variation can indeed change the regulatory networks but how does this impact on phenotypes. Two recent studies looked at particular examples of regulatory changes in S. cerevisiae.
A study from the Barkai lab has linked a change from aerobic to anaerobic growth to the loss of a specific regulatory site in several genes, providing evidence that regularly changes can impact on phenotypes.
To counter this, another study, this time on the mating of yeast has shown that the same phenotype can be observed even if the regulatory mechanism have changed.
These individual examples are just a taste of what we might expect in the future. They do show that regulatory plasticity can be used by the cell to generate phenotypic change but also that selection for phenotype can maintain a function even if the underlying molecular details are not the same.
Tags: